Modelling energy transfer processes
Note: Currently, only one energy transfer process has been implemented, so this section may be subject to change in future releases.
Consider the crystal structure we previously generated. We wish to "dope" that structure with some lanthanide ions randomly around some central donor ion. We will replace our previously defined "central yttrium atom" with a samarium. We will then randomly (based on the concentration of the dopant in the crystal) dope samarium ions around this central samarium ion. This step must be performed many times to "build up" many random unique samarium-samarium donor-acceptor configurations. When the number of random configurations is large enough, this should accurately represent a physical crystal sample. This is the Monte-Carlo aspect of our energy transfer analysis.
To generate our random samples, we utilise the interaction class. This class takes in our structure class and provides additional methods for calculating our interaction components and more specific plotting methods for doped crystals. We can enact this class simply by passing our structure to the interaction class:
crystal_interaction = Interaction(KY3F10)
Generating random dopant configurations is a two-step process. First, we compute the spherical coordinates of all same-species ions within a given radius of the central ion using distance_sim():
crystal_interaction.distance_sim(radius=10)
This populates crystal_interaction.filtered_coords with the coordinates of all candidate sites within the specified radius. These are all Y3+ ions — the sites that could be doped:
print(crystal_interaction.filtered_coords)
r theta phi species
0 7.062817 115.108542 -115.648561 Y3+
1 7.062817 115.108542 -154.351439 Y3+
2 9.855419 55.821457 -135.000000 Y3+
3 7.062817 66.924234 -117.467113 Y3+
4 8.153300 90.000000 -135.000000 Y3+
.. ... ... ... ...
Next, we call doper() to randomly assign dopant species to those sites based on a given concentration (in %) and retrieve the radial distances of the resulting dopant ions:
distances = crystal_interaction.doper(concentration=15, dopant='Sm3+')
By default, this returns a NumPy array of radial distances (in Angstroms) of the dopant samarium ions about the central samarium donor ion:
[8.28402747 7.17592429 7.17592429 9.33368811 8.28402747 4.30030493
8.28402747 7.17592429 9.33368811 7.17592429 3.98372254 8.28402747]
If you want to see the full picture — which sites were doped and their complete spherical coordinates — pass return_coords=True:
doped_config = crystal_interaction.doper(concentration=15, dopant='Sm3+', return_coords=True)
print(doped_config)
r theta phi species
0 7.175924 115.071364 -1.156825e+02 Y3+
1 7.175924 115.071364 -1.543175e+02 Y3+
2 8.284027 90.000000 -1.350000e+02 Y3+
3 7.175924 66.886668 -1.525658e+02 Y3+
4 7.175924 66.886668 -1.174342e+02 Sm3+
5 9.192125 109.317471 -1.800000e+02 Y3+
6 5.861968 92.188550 -1.800000e+02 Y3+
7 9.333688 162.434187 -1.800000e+02 Sm3+
.. ... ... ... ...
This returns a DataFrame showing all candidate sites with their r, theta, phi, and species — where some Y3+ sites have been randomly replaced with Sm3+.
Note that doper() works non-destructively: it temporarily replaces a random subset of Y3+ sites with Sm3+ based on the concentration, extracts the result, and then resets filtered_coords back to the original Y3+ candidate sites. This means filtered_coords is always unchanged after calling doper() — only the returned value contains the result of a particular random doping configuration.
Each call to doper() produces a different random configuration — this is the basis of the Monte Carlo approach.
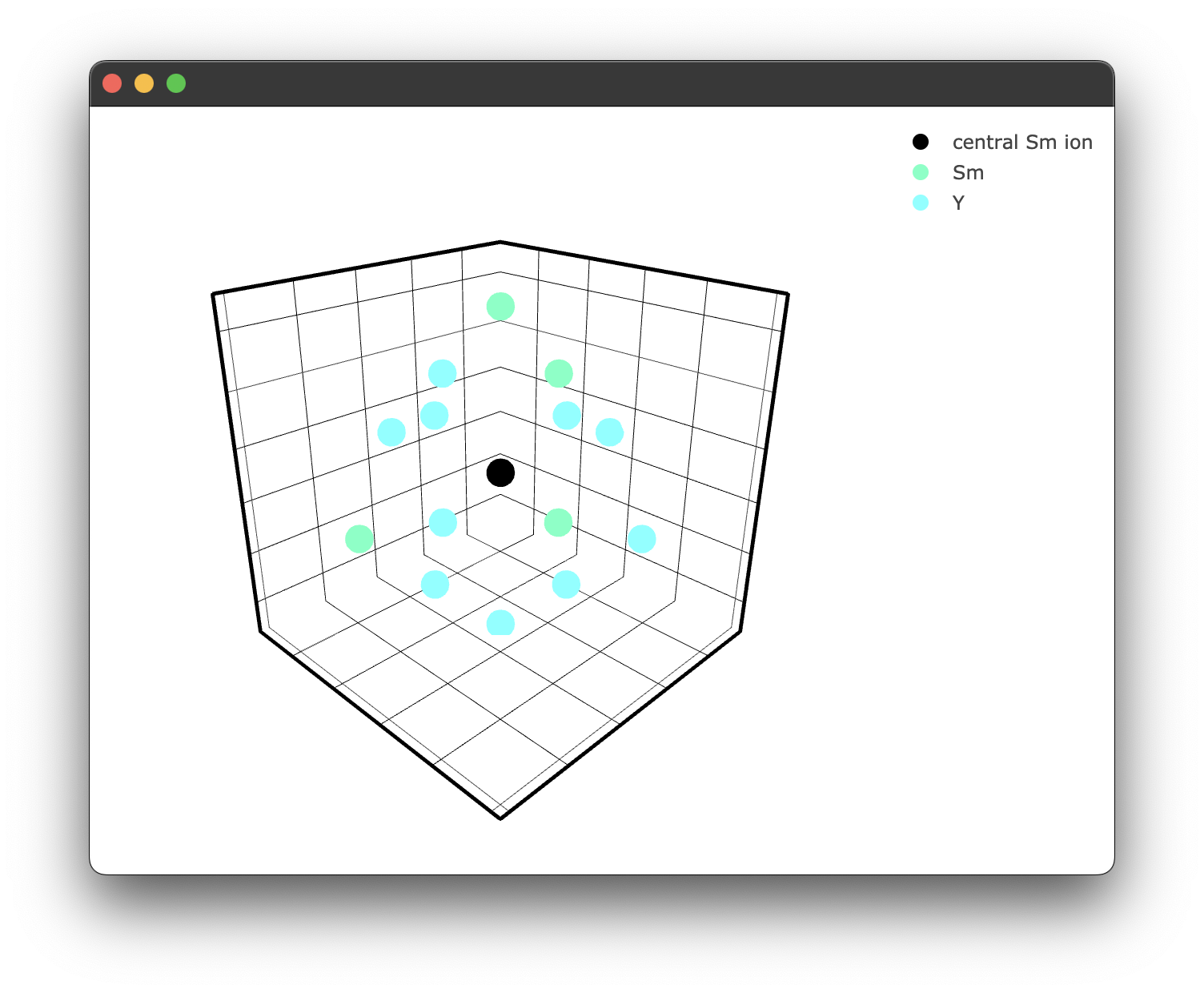
We can also visualise a doped structure to see what is happening; the interaction class has similar plotting functionality.
if __name__ == "__main__":
fig3 = crystal_interaction.doped_structure_plot(radius=7.0, concentration = 15.0 , dopant = 'Sm3+' , filter = ['Y3+','Sm3+'])
fig3.show()
The filter parameter controls which species are shown in the plot. If you omit it, every species in the structure will be plotted. If you provide it, it must be a list of species strings including their charge, e.g. 'Y3+' not 'Y'. Only the species in that list will appear. This is useful for decluttering plots when your structure has many species and you only care about a few of them.
yielding the following figure:
We can now calculate our interaction component for each random doping configuration. This is handled by the sim method, which currently is referred to as sim_single_cross as it is the only implemented method at the time of writing. This method handles both the distance_sim() and doper() steps internally, so you do not need to call them separately when using it. However, it is possible to add your own interaction model by wrapping distance_sim() and doper() for cooperative processes, for example.
interaction_components2pt5pct = crystal_interaction.sim_single_cross(radius=10, concentration = 2.5, interaction_type='DQ', iterations=20)
The sim method takes a radius, concentration, interaction type and number of iterations. The interaction type is given by a two-letter code, i.e. 'DQ' equals dipole-quadrupole. We will need more iterations than just 20 for fitting, closer to 50,000. If we rerun this now with 50,000 iterations, we get the following response:
file found in cache, returning interaction components
This is because I have already run this command, which has been cached. See the notes on caching here. The sim returns a Numpy array of interaction components that matches the number of iterations and will be utilised in our fitting process next!
We can then generate another set of interaction components for a 5% doped sample simply by changing the concentration
interaction_components5pct = crystal_interaction.sim_single_cross(radius=10, concentration = 5, interaction_type='DQ', iterations=50000)
The crystal interaction simulation can also accept the boolean flag intrinsic = True; this uses a modified formulation of the interaction components in the form
This gives us the energy transfer rate () or average energy transfer rate () in terms of a dopant ion 1 Å from the donor ion. The relevance of this is for work regarding an intrinsic energy rate. It is set to false by default as it is not as relevant at this stage; however, in future, it may be of interest.